
Wat is het ICCA-syndroom?
Het ICCA-syndroom is een erfelijk aandoening waarbij kinderen op de babyleeftijd last hebben van epilepsieaanvallen en later tijdens de kinder- en tienerleeftijd last hebben van aanvallen waarbij kinderen ongecontroleerd bewegingen (dyskinesie genoemd) maken.
Hoe wordt het ICCA-syndroom ook wel genoemd?
ICCA is een afkorting voor de woorden Infantiele Convulsies met Chorea Athetose. Convulsies is een ander woord voor epilepsieaanvallen. Het woord infantiel geeft aan dat deze epilepsie aanvallen bij baby’s aanwezig zijn. Chorea en Athetose zijn medische woorden voor de aanvallen van ongecontroleerd bewegen die kinderen op latere leeftijd krijgen.
Benigne familiaire infantiele epilepsie
Een deel van de kinderen heeft alleen op babyleeftijd last van epilepsieaanvallen en krijgt later geen last van aanvallen met ongecontroleerde bewegingen. Deze vorm van epilepsie heeft de naam benigne familiare infantiele epilepsie of beningne infantiele convulsies. Tegenwoordig wordt deze vorm van epilepsie ook wel self-limited familial neonatal-infantile epilepsy genoemd. Het woord self limited geeft aan dat kinderen over deze vorm van epilepsie heen groeien.
Paroxysmale dyskinesie
Er zijn ook kinderen die niet op de babyleeftijd last hebben gehad van epilepsieaanvallen en alleen op latere leeftijd last krijgen van aanvallen met ongecontroleerde bewegingen. Deze aanvallen van ongecontroleerde bewegingen worden paroxysmale dyskinesie genoemd.
Hoe vaak komt het ICCA-syndroom voor bij kinderen?
Het is niet goed bekend hoe vaak het ICCA-syndroom voorkomt bij kinderen. Waarschijnlijk is ook bij lang niet alle kinderen deze diagnose gesteld.

Bij wie komt het ICCA-syndroom voor?
De epilepsie aanvallen die horen bij dit syndroom komen meestal voor bij baby’s en jonge kinderen tussen de leeftijd van 3 en 12 maanden. De aanvallen verdwijnen meestal rond de leeftijd van een jaar. Vanaf de lagere of middelbare schoolleeftijd krijgen kinderen aanvallen waarin ze ongecontroleerde bewegingen maken.
Zowel jongens als meisjes kunnen het ICCA-syndroom hebben.
Wat is de oorzaak van het ICCA-syndroom?
Foutje in het erfelijk materiaal
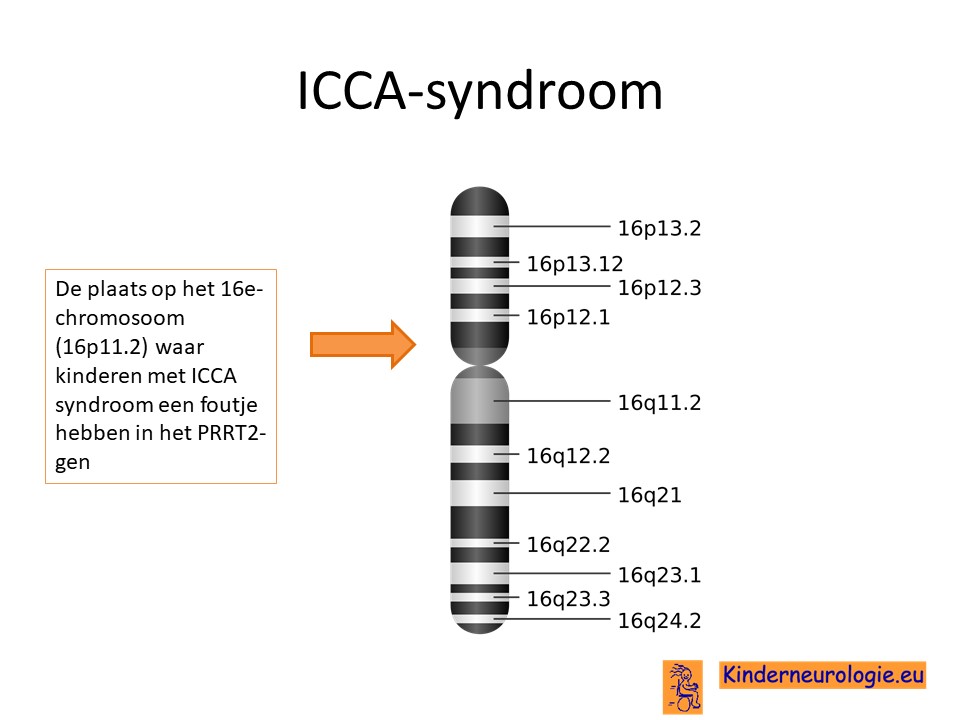
Het ICCA-syndroom wordt veroorzaakt door een fout op een stukje materiaal van het 16e-chromosoom. De plaats van deze fout wordt het PRRT2-gen genoemd.
Waarschijnlijk kunnen een fout op chromosoom 3 en op een andere plek op chromosoom 16 ook zorgen voor het ontstaan van het ICCA-syndroom. Om welke fouten het precies gaat is nog niet bekend.
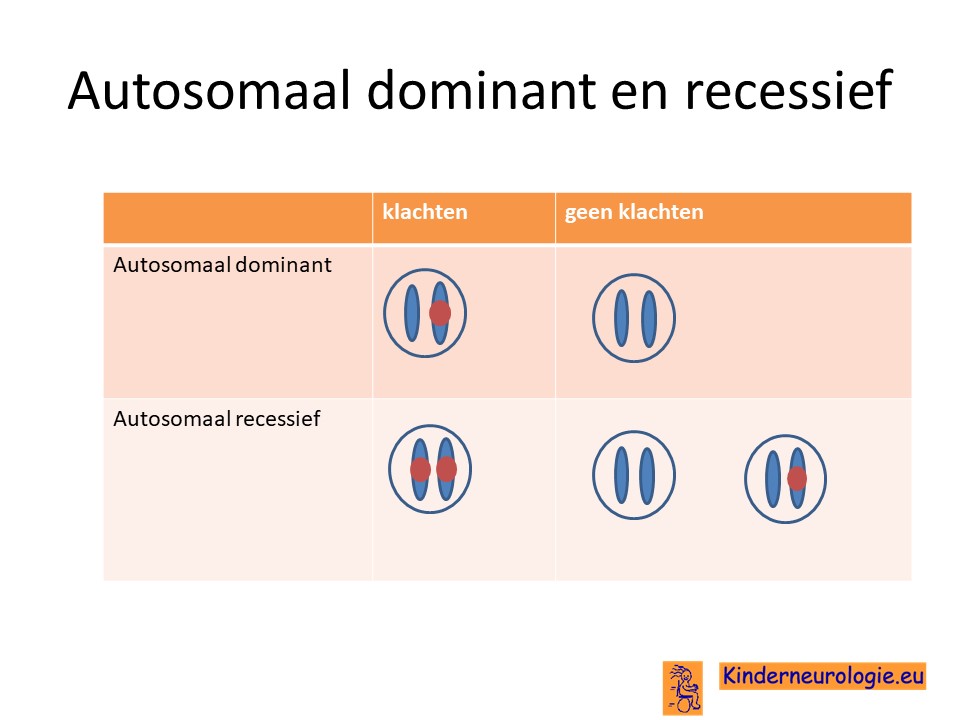
Autosomaal dominant
Het ICCA-syndroom wordt veroorzaakt door een zogenaamde autosomaal dominante fout. Dit houdt in dat een fout op een van de twee chromosomen 16 die een kind heeft in het PRRT2-gen al voldoende is om de aandoening te krijgen. Dit in tegenstelling tot een autosomaal recessieve fout waarbij kinderen pas klachten krijgen wanneer beide chromosomen een fout bevatten.
Geërfd van een ouder
Een deel van de kinderen heeft de fout in het PRRT2-gen geërfd van een ouder die zelf ook een fout in het PRRT2-gen heeft. Soms was dit al bekend, soms wordt de diagnose bij de ouder pas gesteld wanneer bij het kind de diagnose gesteld wordt.

Bij het kind zelf ontstaan
Bij een andere deel van de kinderen met een ICCA-syndroom is het foutje bij het kind zelf ontstaan na de bevruchting van de eicel door de zaadcel en niet overgeërfd van een van de ouders. Dit wordt de novo genoemd, wat nieuw ontstaan betekent. Het kind is dan de eerste in de familie die deze aandoening heeft.

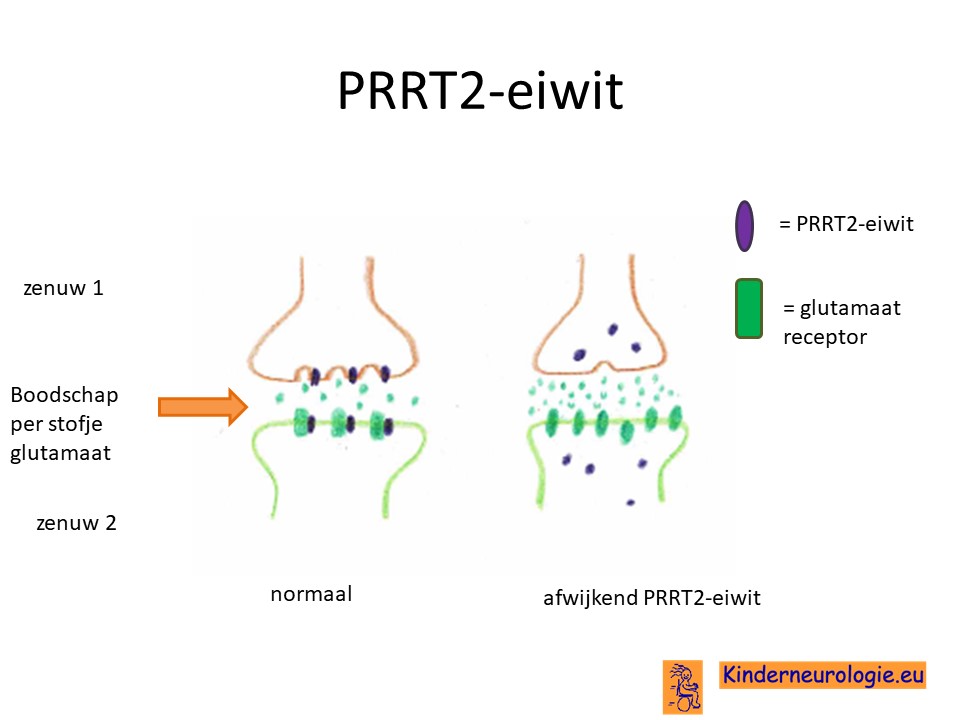
Afwijkend eiwit
Dit stukje chromosoom bevat informatie voor de aanmaak van een eiwit, PRRT2-eiwit genoemd. PRRT2 staat voor Proline Rich Transmembraan Proteine 2. Dit eiwit speelt een belangrijke rol in de wand van hersencellen op de plaats waar twee hersencellen met elkaar communiceren. Zo’n plaats waar twee hersencellen met elkaar communiceren wordt een synaps genoemd. Het PRRT2-gen zorgt er voor dat de boodschapperstofjes goed worden afgegeven in de ruimte tussen twee zenuwen. Zonder goed werkend PRRT2 worden de boodschapperstofjes niet in juiste hoeveelheid afgegeven. Op bepaalde momenten te veel van het boodschapper stofje glutamaat afgegeven waardoor epilepsie of overmatige beweeglijkheid ontstaan. Bij de overmarige beweeglijkheid blijken vooral hersencellen in de kleine hersenen niet goed te functioneren.
Wat zijn de symptomen van het ICCA-syndroom?
Variatie
Er bestaat variatie in de hoeveelheid en in de ersnt van de symptomen die verschillende kinderen met het ICCA-syndroom hebben. Sommige kinderen hebben vooral last van de epilepsieaanvallen, andere kinderen van de bewegingsproblemen en weer andere kinderen beide problemen in gelijke mate.

Epilepsie
Een groot deel van de kinderen met het ICCA-syndroom krijgt epilepsieaanvallen op de baby en dreumesleeftijd. Vaak beginnen de aanvallen tussen de leeftijd van drie en twaalf maanden.
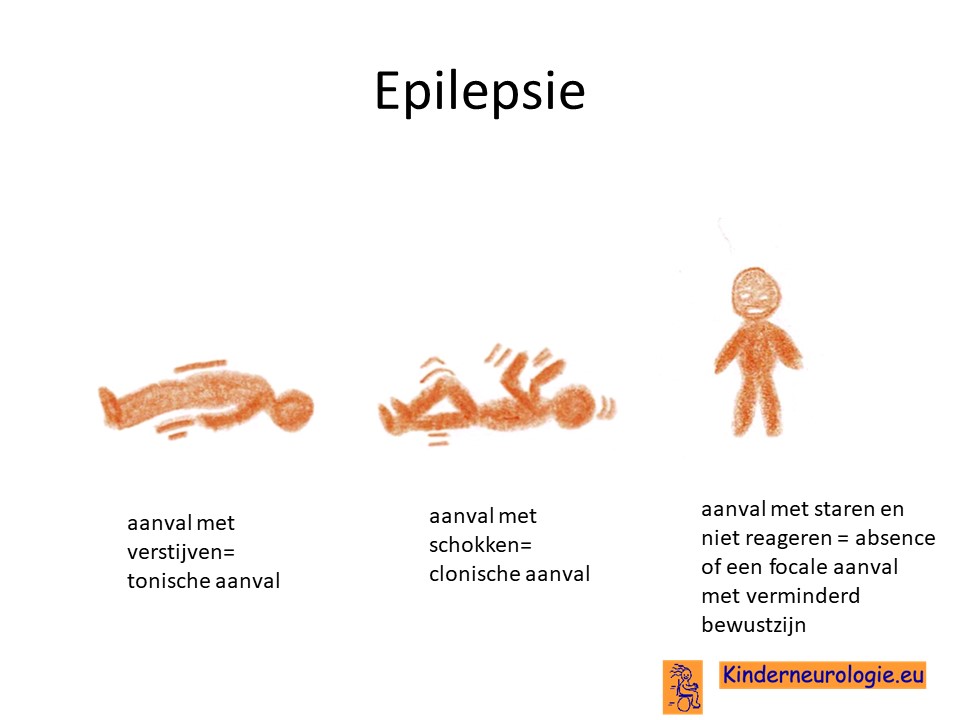
Focale aanvallen
Tijdens de aanvalletjes houden kinderen plotseling op met dat waarmee ze bezig waren, de kinderen gaan staren, reageren niet meer goed op de omgeving, soms draaien de ogen en het hoofd naar een kant toe en vaak zijn er enkele schokjes aan een kant van het lichaam. Soms zit de aanval aan de ene kant van het lichaam, de andere keer zit de aanval aan de andere kant van het lichaam. Een enkele keer breiden de schokken zich uit naar twee kanten van het lichaam.
Ontwikkeling
Kinderen met het ICCA-syndroom ontwikkelen zich normaal. Sommige kinderen ontwikkelen zich in de eerste maanden wel wat trager dan leeftijdgenootjes zonder aanvallen.
Maar dit verdwijnt wanneer de aanvallen verdwijnen.
Geen klachten
Na verdwijnen van de epilepsieaanvallen volgen een aantal jaren waarin er geen bijzonderheden zijn bij kinderen met het ICCA-syndroom ten opzichte van leeftijdsgenoten.
Aanvallen met ongecontroleerde bewegingen
Vanaf de lagere schoolleeftijd (varierend van de leeftijd van 3 tot 12 jaar) krijgen kinderen aanvallen waarin zij gedurende enkele minuten ongecontroleerde bewegingen maken met hun handen of met hun benen. Deze bewegingen worden ook wel chorea of dystonie genoemd. Deze aanvallen worden nog al eens uitgelokt door plotseling in beweging komen of door plotseling schrikken. In periodes met stress of angst hebben kinderen meer last van deze aanvallen. Kinderen hebben vaak meerdere aanvallen op een dag. Tijdens de aanvallen zijn kinderen gewoon bij bewustzijn. Zij kunnen echter geen controle uitoefenen op de bewegingen die ze maken. De aanvallen verdwijnen meestal weer op jong volwassen leeftijd.

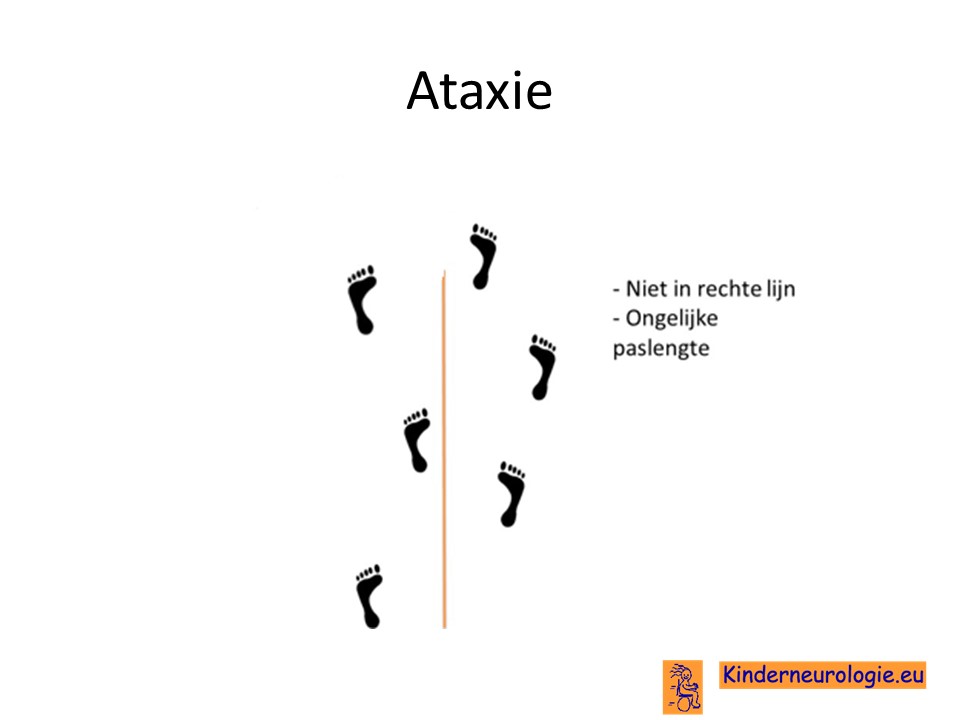
Problemen met het evenwicht
Er zijn ook kinderen die tijdens de aanval last krijgen van problemen met het bewaren van de balans. Dit wordt ook wel episodische ataxie genoemd.
Tics
Kinderen met ICCA-syndroom kunnen ook last hebben van tics. Tics zijn korte snelle onwillekeurige bewegingen in het gezicht of in de handen. Tics kunnen wel kortdurend worden tegen gehouden door kinderen, maar dit geeft wel een heel vervelend gevoel. Wanneer de tics een tijdje tegen gehouden zijn, dan komen ze er daarna vaak tijdelijk even nog vaker voor. De tics moeten er als het ware uit.
Migraine
Kinderen met het ICCA-syndroom hebben vaker last van migraineaanvallen. Migraine is een hoofdpijnaanval waarbij kinderen last hebben van bonzende hoofdpijn in het hele hoofd of in een deel van het hoofd. Tijdens de hoofdpijnaanvallen hebben kinderen vaak last van misselijkheid en kunnen kinderen gemakkelijk gaan spugen. Licht en geluid zijn vaak vervelend. Tijdens de migraine aanval kunnen kinderen tijdelijk last hebben van een verlamming of een gevoelsstoornis in een arm of in een been. Dit verdwijnt weer als de migraine aanval voor bij is.
Problemen met leren
Een deel van de kinderen met het ICCA-syndroom heeft problemen met leren. Zij hebben meer moeite om langere tijd geconcentreerd met hun werk bezig te zijn. Kinderen kunnen het moeilijker vinden om het geleerde in nieuwe situaties toe te passen.
Hoe wordt de diagnose ICCA-syndroom gesteld?
Verhaal en onderzoek
De diagnose ICCA-syndroom kan worden vermoed op grond van het verhaal van een kind die op babyleeftijd epilepsieaanvallen heeft en later op kinderleeftijd aanvallen krijgt met onwillekeurige bewegingen.
Wanneer in een familie al bekend is dat meerdere mensen het ICCA-syndroom hebben, dan zal de diagnose gemakkelijk te stellen zijn.
EEG
Bij baby’s met epilepsieaanvallen als gevolg van een ICCA-syndroom zal vaak een EEG gemaakt worden. Wanneer een baby tijdens het EEG geen aanvalletjes krijgt, dan ziet het EEG er volkomen normaal uit. Wanneer het wel zo is dat een baby een aanval krijgt tijdens het EEG dan wordt er epileptiforme activiteit gezien in de vorm van pieken in een bepaald gebied van de hersenen. Dit gebied kan per aanval wisselen en ook variëren tussen de rechter en de linkerkant van de hersenen. De epileptiforme activiteit kan tijdens een aanval uitbreiden naar de gehele hersenen.
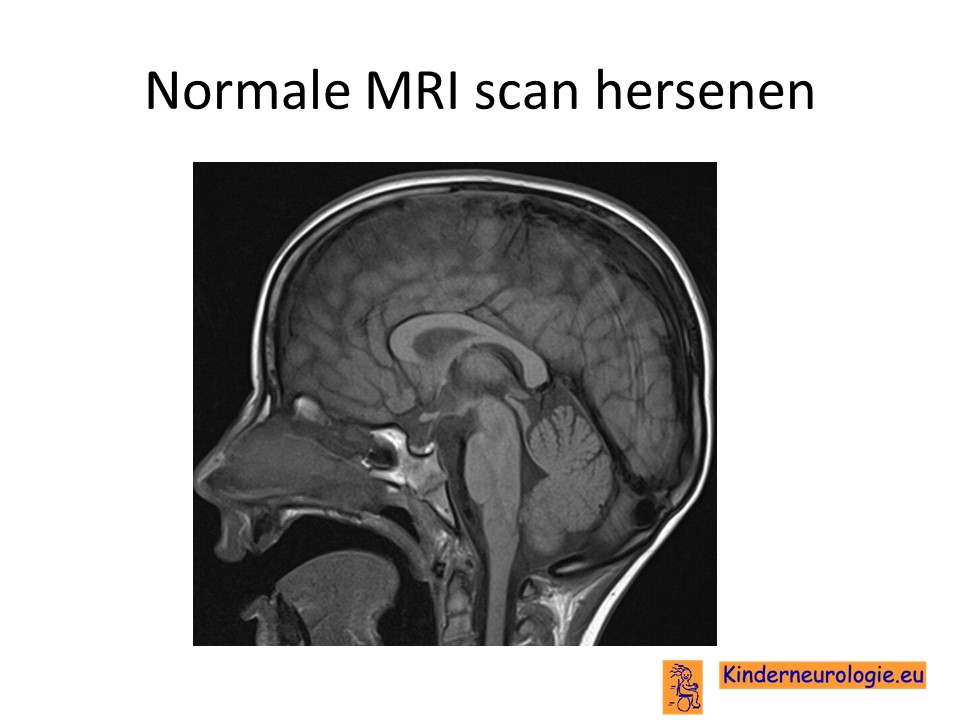
MRI-scan
Bij baby’s met epilepsieaanvallen waarbij nog niet bekend is dat dit als gevolg van het ICCA-syndroom is zal vaak een MRI scan van de hersenen gemaakt worden. Op deze MRI scan worden bij kinderen met het ICCA-syndroom geen afwijkingen gezien.
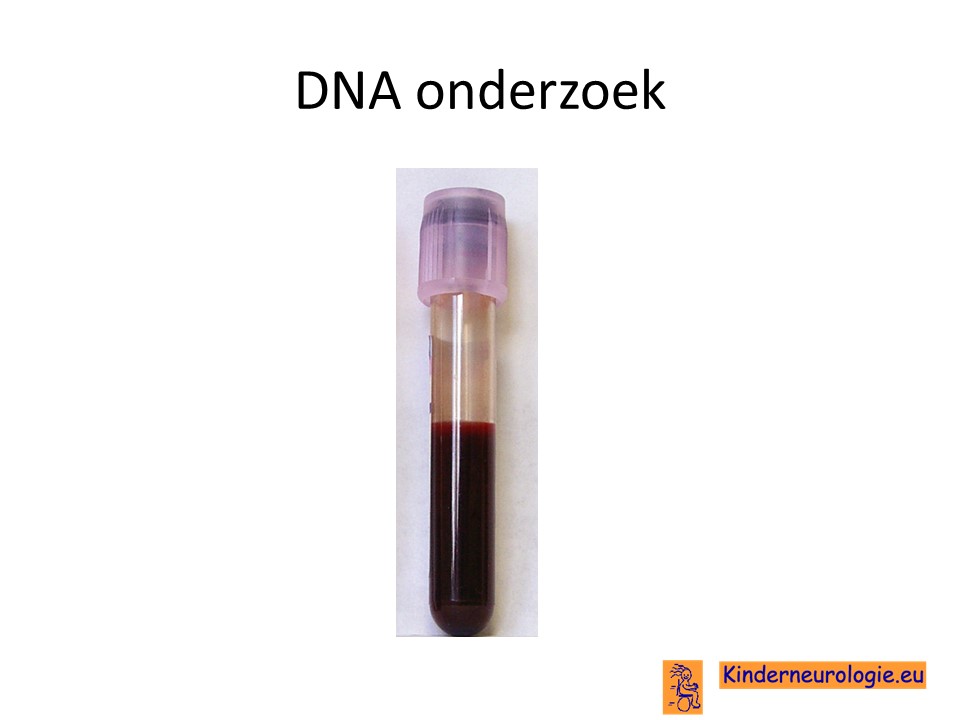
Genetisch onderzoek
Wanneer aan het ICCA-syndroom wordt gedacht kan door middel van bloedonderzoek de fout in het erfelijk materiaal van het 16e chromosoom in het ICCA-gen worden aangetoond.
Vaak worden ook alle chromosomen tegelijkertijd onderzocht (zogenaamd Array onderzoek), meestal kan niet op deze manier de diagnose ICCA-syndroom worden gesteld.
In de toekomst zal door middel van een nieuwe genetische techniek (exome sequencing genoemd) mogelijk ook deze diagnose gesteld kunnen worden zonder dat er specifiek aan gedacht was of naar gezocht is.
Stofwisselingsonderzoek
Wanneer jonge kinderen epilepsie aanvalletjes krijgen, zal vaak gekeken worden of er sprake is van een stofwisselingsziekte. Bij dit onderzoek worden bij kinderen met het ICCA-syndroom geen afwijkingen gevonden.
Hoe wordt het ICCA-syndroom behandeld?
Geen behandeling
Niet alle kinderen met epilepsie als gevolg van het ICCA-syndroom hebben een behandeling nodig. Het hangt af van de ernst van de aanvallen en de frequentie van de aanvallen of de voordelen van het krijgen van medicijnen opwegen tegen de nadelen van het krijgen van medicijnen.
Aanvalsbehandeling epilepsie
De meeste epilepsieaanvallen gaan vanzelf over binnen enkele minuten. Omstanders hoeven dan niets te doen om de aanval te doen stoppen. Het is belangrijk om zo rustig mogelijk te blijven en het kind zo veel mogelijk met rust te laten.
Wanneer een aanval na 3-5 minuten nog niet vanzelf gestopt is, dan zal vaak geadviseerd worden om medicijnen te geven om een aanval te doen stoppen. De behandelende arts zal altijd aangeven welk tijdstip voor een bepaald kind het beste is. Medicijnen die gebruikt kunnen worden voor het stoppen van een aanval zijn diazepam rectiole (Stesolid®), midazolam neusspray, midazolam rectiole, lorazepam of clonazepam druppels.
Het effect van deze medicijnen ontstaat na enkele minuten. Nadien zal het kind meestal in slaap vallen, soms ook niet en vertonen kinderen juist druk gedrag na geven van deze medicatie.

Medicijnen om nieuwe epilepsie aanvallen te voorkomen
Wanneer medicatie nodig is omdat de epilepsie aanvallen frequent voorkomen, kunnen verschillende soorten medicijnen gebruikt worden zoals fenobarbital (Luminal®), levetiracetam (Keppra®) of carbamazepine (Tegretol ®). Maar ook andere medicijnen die epileptische aanvallen kunnen voorkomen kunnen gebruikt worden. Meestal worden deze medicijnen gegeven totdat gedurende een jaar geen aanvallen meer worden gezien. Dan wordt geprobeerd of het lukt om de medicijnen af te bouwen.

Medicijnen om aanvallen van bewegingsstoornis te voorkomen
Carbamazepine (Tegretol ®), levetiracetam (Keppra®) en difantoine (Fenytoine ®) kunnen ook helpen om aanvallen van ongecontroleerde bewegingen als gevolg van het ICCA-syndroom te voorkomen. Vaak is maar een lage dosering van deze medicijnen nodig om te zorgen dat de aanvallen van dyskinesie niet meer voorkomen.
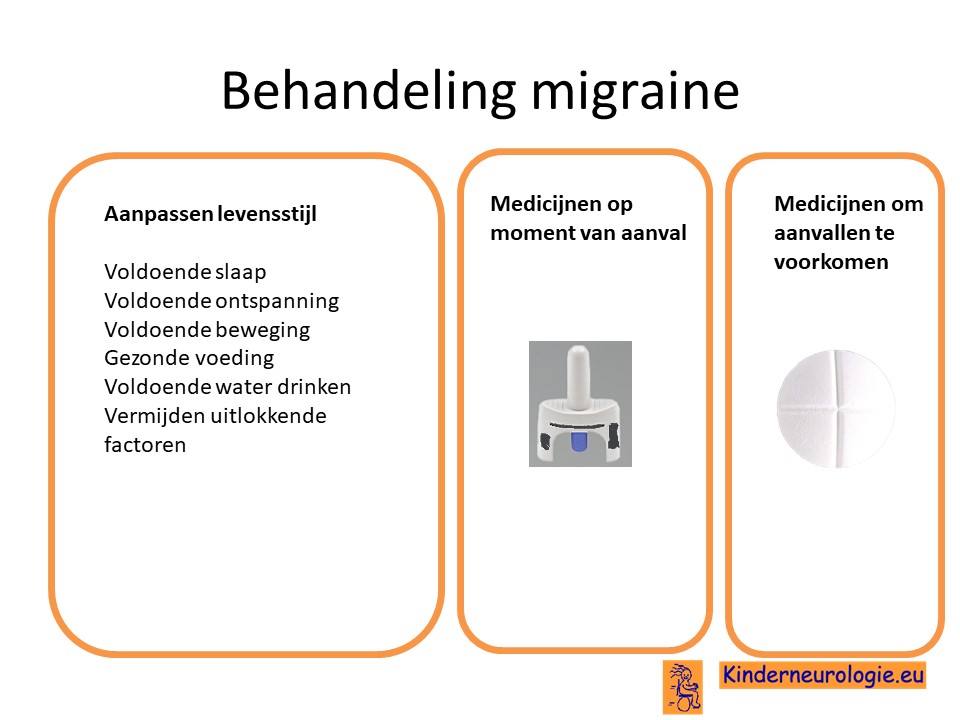
Migraine
Kinderen met ICCA-syndroom die last hebben van migraine aanvallen, worden op dezelfde manier behandeld als kinderen met migraine zonder het ICCA-syndroom.
Begeleiding
Een maatschappelijk werkende of psycholoog kan begeleiding geven hoe het hebben van deze aandoening een plaatsje kan krijgen in het dagelijks leven. Het kost vaak tijd voor ouders om te verwerken dat hun kind een neurologische aandoening heeft.

Contact met andere ouders
Door middel van een oproepje op het forum van deze site kunt u proberen in contact te komen met andere kinderen en hun ouders/verzorgers die ook te maken hebben met het ICCA-syndroom.
Wat betekent het hebben van het ICCA-syndroom voor de toekomst?
Verdwijnen
Bij de meeste kinderen met epilepsieaanvallen als gevolg van het ICCA-syndroom verdwijnen de aanvallen vanzelf rond de leeftijd van één tot twee jaar. Een enkele keer houden de epilepsieaanvallen aan tot de leeftijd van drie jaar.
Ook de aanvallen met onwillekeurige bewegingen verdwijnen meestal weer tegen het einde van de puberteit/ jong volwassen leeftijd.
Normale ontwikkeling
Kinderen met het ICCA-syndroom ontwikkelen zich normaal. Zij kunnen later zelfstandig functioneren in de maatschappij.
Transitie van zorg
Tussen de leeftijd van 16 en 18 jaar wordt de zorg vaak overgedragen van kinderspecialisten naar specialisten die de zorg aan volwassenen geven. Het is belangrijk om tijdig hierover na te denken. Is er behoefte de zorg over te dragen naar specialisten voor volwassenen of kan de huisarts de zorg leveren die nodig is.En als er behoefte is aan overdragen van de zorg naar specialisten voor volwassenen, naar welke dokter(s) wordt de zorg dan overgedragen? In welk ziekenhuis kan de zorg het beste geleverd worden. Het proces van overdragen van de zorg wordt transitie genoemd. Het is belanrgijk hier tijdig over na te denken en een plan voor te maken samen met de dokters die betrokken zijn bij de zorg op de kinderleeftijd.
Ook verandert er veel in de zorg wanneer een jongere de leeftijd van 18 jaar bereikt. Voor meer informatie over deze veranderingen verwijzing wij u naar het artikel veranderingen in de zorg 18+.
![]()
Levensverwachting
Kinderen met het ICCA-syndroom hebben een normale levensverwachting.
Kinderen
Volwassenen met het ICCA-syndroom kunnen normaal kinderen krijgen. Deze kinderen hebben tot maximaal 50% kans om zelf ook het ICCA-syndroom te krijgen. Wanneer een kind de fout erft van een van de ouders, betekent dit nog niet dat dit kind ook het ICCA-syndroom gaat krijgen.
Andere vormen van epilepsie
Hoewel de epilepsie aanvallen als gevolg van het ICCA-syndroom met het ouder worden, zullen verdwijnen, blijven kinderen wel gevoeliger voor het krijgen van ander type epilepsie aanvalletn. Een op de vijf tot tien kinderen krijgt een andere epilepsie vorm op de kinderleeftijd.
Hebben broertjes en zusjes een verhoogde kans om ook het ICCA-syndroom te krijgen?
Het ICCA-syndroom is een erfelijke aandoening. Wanneer een van de ouders zelf het ICCA-syndroom heeft gehad, dan hebben broertjes en zusjes tot 50% kans om zelf ook het ICCA-syndroom te krijgen.
Wanneer de fout in het erfelijk materiaal bij het kind zelf is ontstaan, dan is de kans heel klein dat broertjes en zusjes zelf ook het ICCA-syndroom krijgen. Dit zou alleen het geval zijn wanneer een van de ouders het foutje in het PRRT2-gen in de eicel of in de zaadcel heeft zitten, zonder dat het in de andere lichaamscellen zitten. De kans daarop is heel klein.
Een klinisch geneticus kan hier meer informatie over geven.

Familiebrief
Het hebben van een genetische aandoening kan soms ook consequenties hebben voor andere familieleden dan alleen de jongere en zijn gezin. Er kan een kans bestaan dat deze aandoening bij meerdere familieleden voorkomt. Een klinisch geneticus maakt meestal een familiebrief. Hierin wordt uitgelegd wat de aandoening inhoudt, waar meer informatie te vinden is over de aandoening en waarin vermeld staat of familieleden een verhoogde kans hebben om ook zelf deze aandoening te hebben. Met deze brief kunnen familieleden die dat willen via de huisarts verwezen worden naar een klinisch geneticus.
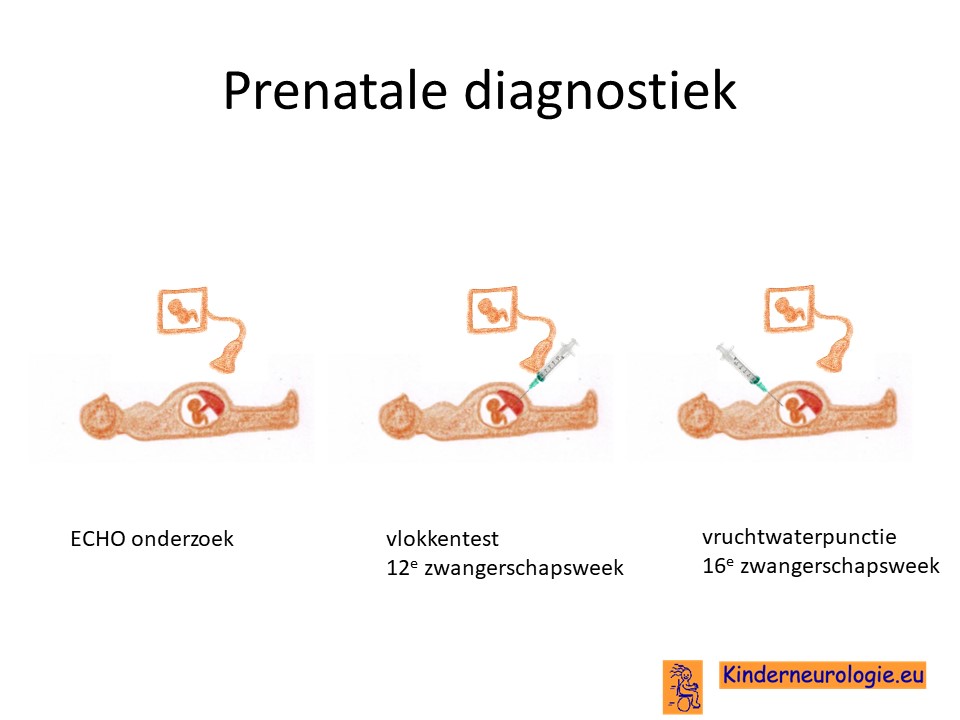
Prenatale diagnostiek
Het is mogelijk om tijdens een volgende zwangerschap prenatale diagnostiek te verrichten in de vorm van een vlokkentest in de 12e zwangerschapsweek of een vruchtwaterpunctie in de 16e zwangerschapsweek. Zo kan gekeken worden of dit kindje ook het ICCA-syndroom heeft. Beide ingrepen hebben een klein risico op het ontstaan van een miskraam (0,5% bij de vlokkentest en 0,3% bij de vruchtwaterpunctie). De uitslag van deze onderzoeken duurt twee weken. Voor prenatale diagnostiek kan een zwangere de 8ste week verwezen worden door de huisarts of verloskundige naar een afdeling klinische genetica. Meer informatie over prenatale diagnostiek kunt u vinden op de website: www.pns.nl
Wilt u dit document printen dan kunt u hier een pdf-versie downloaden.
Wilt u ook uw verhaal kwijt, dat kan: verhalen kunnen gemaild worden via info@kinderneurologie.eu en zullen daarna zo spoedig mogelijk op de site worden geplaatst. Voor meer informatie zie hier
Heeft u foto's die bepaalde kenmerken van deze aandoening duidelijk maken en die hier op de website mogen worden geplaatst, dan vernemen wij dit graag.
Links en verwijzingen
www.epilepsievereniging.nl
(site van epilepsievereniging Nederland)
www.epilepsie.nl
(site van het nationaal epilepsiefonds)
Referenties
Laatst bijgewerkt 2 maart 2019, voorheen: 12 januari 2016
Auteur: JH Schieving
Heeft uw kind nog andere symptomen, laat het ons weten.